Article overview: A recent paper proposed that muscle weakness in severe ME/CFS may be driven by dysfunction of the sodium-potassium pump in skeletal muscle cells. The authors argue that this could disrupt cellular electrical signalling and cause sodium and calcium overload, which in turn impairs mitochondria, and contributes to the clinical threshold for post-exertional malaise (PEM). Whilst alternative explanations such as deconditioning and muscle damage are discussed, the paper presents an electrophysiological model centred on ion imbalance as a unifying mechanism – though this remains a hypothesis requiring further research.
Muscle weakness in severe ME/CFS: If not the nerves, then what?
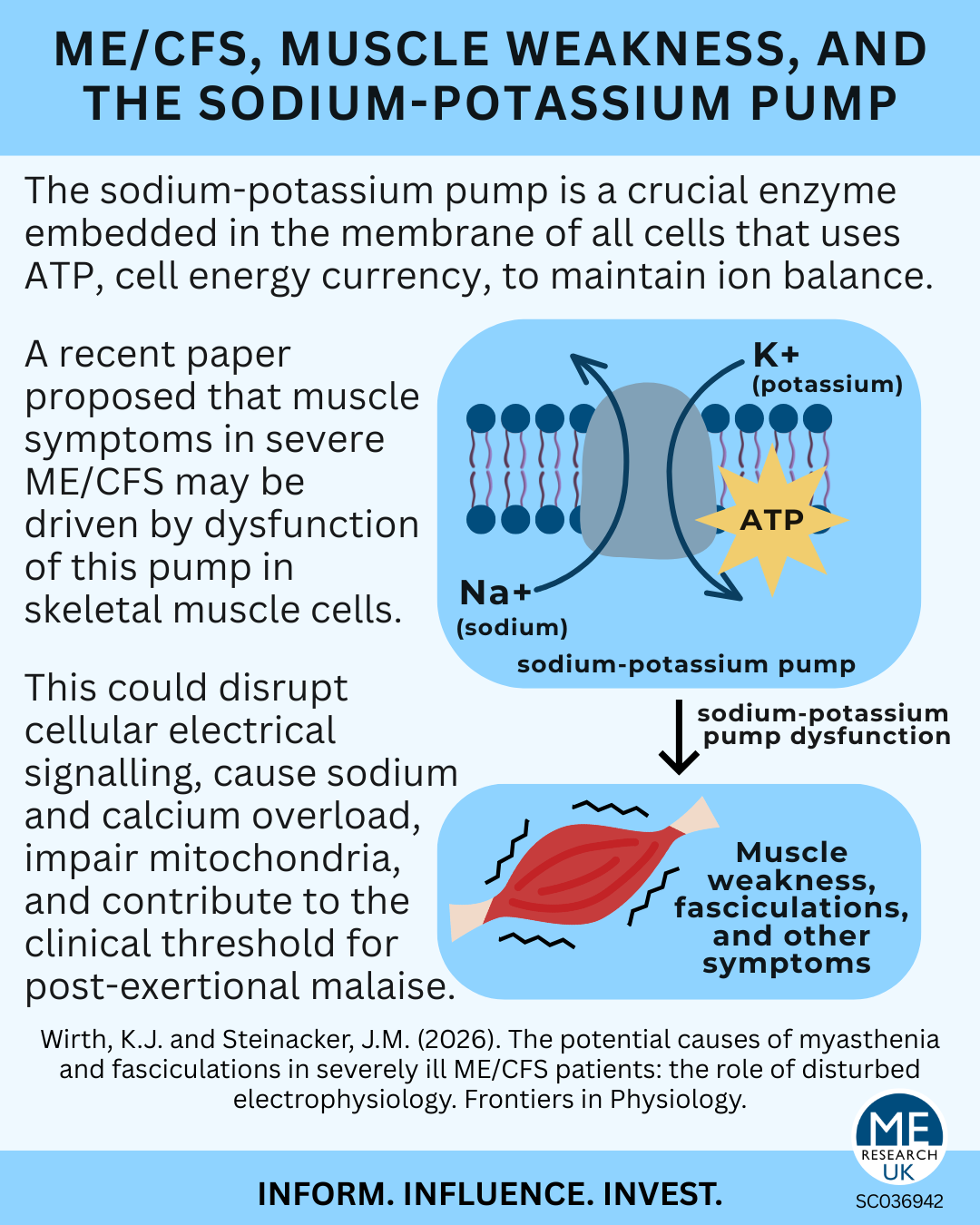
In a recent paper, Prof. Dr Klaus Wirth and Prof. Dr Jürgen Steinacker explore the causes of skeletal muscle (muscles connected to bone enabling movement) symptoms including, loss of force, fatigue, pain, fasciculations (twitches/tremor) in individuals with severe ME/CFS.
They highlight that myasthenia (muscle weakness) contributes to disease burden and can be characterised by an “inability to perform even low-intensity endurance exercises”. They note reduced hand grip strength correlates with greater severity of disability, fatigue, and other symptoms.
Importantly, the researchers state that neurological investigations exclude neuronal (nerve-related) causes of myasthenia (muscle weakness), hence they looked elsewhere for clues about what is happening.
Wirth and Steinacker propose an alternative explanation centred on a fundamental cellular element: the sodium-potassium pump.
The majestic sodium-potassium pump
The sodium-potassium pump (Na⁺/K⁺-ATPase) is an enzyme embedded in the membrane of all cells. Using ATP (the cell’s energy currency), it moves sodium out of the cell and potassium into it. This process maintains the cell’s resting membrane potential – essential for conducting electrical impulses along nerves and enabling muscle contraction.
The authors hypothesise that the sodium potassium pump is dysfunctional in the skeletal muscle of severely ill individuals with ME/CFS leading to downstream effects and eventually muscle issues.
Although the paper argues that the issue is not neuronal, it is worth noting that the sodium-potassium pump plays a central role in nerve conduction. Thus, even if nerves are structurally intact, impaired membrane electrophysiology could still disrupt neuromuscular (nerve-muscle) function.
Theory against theory
The authors contextualise their electrophysiological hypothesis by examining – and challenging – three alternative explanations for muscle weakness:
1. Lack of energy due to mitochondrial dysfunction
Mitochondria generate cellular energy. Whilst mitochondrial dysfunction could explain reduced endurance, the authors argue it cannot account for a marked loss of force during the very first contraction after adequate rest.
Some skeletal muscle fibres are glycolytic, meaning they rely less on mitochondria during initial activity. Therefore, mitochondrial impairment alone should not produce immediate weakness. That said, the paper goes on to indicate that mitochondrial dysfunction may still play a role in the disease process.
2. Deconditioning and atrophy (wasting) due to inactivity in the intention to avoid post-exertional malaise
The authors argue against this. They cite research comparing healthy individuals after six weeks of bed rest with patients with ME/CFS and long COVID. Unlike bed-rested healthy controls, individuals with ME/CFS did not show muscle atrophy. Rather, skeletal muscle in ME/CFS may not “rest” fully due to individuals suffering cramps and fasciculations.
They further hypothesise that elevated intracellular calcium levels could simultaneously contribute to muscle damage whilst noting note cellular pathways involving calcium are associated with growth processes that oppose the tendency for atrophy.
3. Structural skeletal muscle damage
Although some research shows skeletal muscle damage in ME/CFS, the authors argue that the extent observed so far may not be sufficient to explain severe loss of force. They suggest muscle biopsies from severely ill patients are needed for clarification.
A disturbed electrophysiology
Wirth and Steinacker argue that “the main and common cause of both loss of force and fasciculations could be a disturbed electrophysiology of skeletal muscle.” Nevertheless, it is important to recognise that this is a hypothesis, that would require further investigation.
Seemingly to illustrate the importance of the sodium-potassium pump, they reference ouabain, a compound that blocks the pump. When applied to isolated skeletal muscle, ouabain causes depolarisation and loss of force upon stimulation. Muscle cells become inexcitable and unable to respond properly – resulting in weakness or even paralysis.
The analogy underscores how crucial proper pump function is to muscle contraction.
Sodium overload leading to calcium overload leading to post-exertional malaise
If the sodium-potassium pump is faulty in the skeletal muscles of individuals with ME/CFS then:
- It can’t pump out sodium as well thus the inside of the cell becomes overloaded with sodium. This compounded by other effects in post-acute infectious syndromes (such as ME/CFS) leads to excessive sodium within cells.
- Excessive intracellular (inside cell) sodium means that the sodium-calcium exchanger, another cell component, reverses direction and starts moving sodium out of cell and moving in calcium.
- As there is so much sodium to move out, an excessive amount of calcium will be brought in (calcium overload), which can lead to mitochondrial damage.
The researchers suggest that the threshold at which the sodium-calcium exchanger reverses could be the biological basis for the “clinical PEM threshold”. They argue that the described disease mechanisms could also explain exercise-induced muscle damage.
Autoantibodies targeting the pump
Stimulating beta-2(ß2) adrenergic receptors, found on the cell membranes of various cells, increases sodium-potassium pump activity. The authors state -“Autoantibodies against [beta-2] adrenergic receptors and the high tendency for [desensitisation] of this receptor by the high sympathetic tone found in ME/CFS” may cause dysfunction of the sodium-potassium pump.
Reactive oxygen species
Although the authors seem to challenge mitochondrial dysfunction as the primary driver of muscle symptoms, they state that mitochondrial dysfunction develops in established ME/CFS.
Reactive oxygen species (ROS) produced due to dysfunctional mitochondria can even inhibit the sodium-potassium pump.
Since the pump has high energy demands, any ATP shortage (as a result of dysfunctional mitochondria) further compromises its activity.
Note: One of the authors was employed by a company focused on developing oral therapeutics targeted at mitochondrial dysfunction. This was transparently declared as a conflict of interest. Whilst worth bearing in mind, this statement is not to imply that the role influenced the paper.
Insulin resistance
Insulin stimulates the sodium-potassium pump. The authors mention that insulin resistance (impaired response to insulin), likely mild, has been reported in individuals with ME/CFS. They posit that in ME/CFS insufficient stimulation by insulin may mean that the skeletal muscle does not sufficiently recover from previous muscle work for the next activity. Patients may start muscle activity under unfavourable conditions, which lead to an early increase in intracellular sodium and calcium.
This could contribute to a low PEM threshold described in their model.
Increased muscle tone leading to fasciculations
The sarcolemma (muscle cell membrane) may be chronically depolarised in ME/CFS, positioning it closer to the threshold for firing an action potential (electrical spark required for a muscle to move). This could increase central muscle tone (ongoing tension within skeletal muscle maintained by central nervous system) and promote hyperexcitability. This manifests as muscle fasciculations and cramps.
Potential contributors include dysfunction of the crucial TRPM3 ion channel and autoantibodies targeting other cell components (SSRM3 protein and alpha2C-adrenergic receptor). Frequent excitations increase the workload of the already impaired sodium-potassium pump, consuming large amounts of ATP even at rest.
This contributes to:
- Increased production of lactate (metabolic byproduct that builds up in muscle during exercise making them ‘burn’)
- Rising intracellular sodium
- Calcium overload
- Mitochondrial damage
Thus, a vicious cycle emerges.
Systemic spillover effects
Energy-depleted muscle may release vasoactive mediators such as bradykinin. If produced excessively, these mediators may spill into systemic circulation and contribute to broader symptoms/signs – including pain, oedema, and spasms – through inflammatory and vascular effects.
Conclusion
Wirth and Steinacker conclude that a dysfunctional sodium-potassium pump could represent a central mechanism underlying muscle symptoms in severe ME/CFS. In their model, impaired pump function leads to intracellular sodium accumulation, causing calcium overload via reversal of the sodium-calcium exchanger. Elevated intracellular calcium may then damage mitochondria and disrupt key cellular processes in skeletal muscle, leading to lack of energy, fatigue, and loss of endurance.
At the same time, disturbed membrane electrophysiology may result in chronic depolarisation of the sarcolemma (muscle fibre membrane). This could impair electrical signalling and muscle fibre recruitment (needed for movement), explaining both loss of force and hyperexcitability manifesting as fasciculations and cramps.
Whilst the paper presents a compelling and interesting theory, clearer framing of competing models earlier in the text may have reduced potential confusion for non-specialist readers. Notably, although the authors argue against lack of energy due to mitochondrial dysfunction playing a primary role in loss of muscle force, mitochondrial dysfunction remains embedded within their own electrophysiological framework. Nevertheless, the paper provides a thought-provoking overview of skeletal muscle abnormalities in ME/CFS, and offers a noteworthy perspective.
Research does not always have an immediate, tangible impact, but it can help guide future understanding and potential treatments, and validate patient experiences. This paper proposes a biological mechanism that may help explain muscle symptoms and PEM in ME/CFS and potentially inform future therapeutic research.